當地時間6月13日,美國(guó)FDA發布了“器械軟件功能(néng)上市前提交内容”的最新(xīn)指南,該指南取代了2005年發布的版本,并更新(xīn)了FDA推薦申辦(bàn)方在上市前提交用(yòng)于對器械軟件功能(néng)進行審查的文(wén)檔,基于風險的方法的想法。
本指南中(zhōng)的建議旨在促進FDA的上市前審查。本指南描述了在軟件開發、驗證和驗證期間通常會生成和記錄的信息。根據FDA的經驗,采用(yòng)了最少負擔的方法來确定通常需要的最低信息量,以支持使用(yòng)軟件器械的上市前提交。
申辦(bàn)方在上市前提交的文(wén)件中(zhōng)納入FDA對軟件功能(néng)器械的安(ān)全性和有(yǒu)效性的評估。這種想法認可(kě)了《21st Century Cures Act (Cures Act)》最近對《FD&C法》所做的修改,它修訂了FD&C法案的第520條,并将某些軟件功能(néng)排除在設備定義之外。它還考慮了數字健康的快速發展的本質(zhì),以及最近FDA認可(kě)的與軟件相關的共識标準。
本指南不适用(yòng)于評估上市後軟件器械問題時可(kě)能(néng)需要的軟件相關文(wén)檔,包括更正和删除。
雖然本指南确定了申辦(bàn)方應在上市前提交中(zhōng)包含的文(wén)件,但本指南并不打算提供關于如何開發、驗證和驗證設備軟件的建議。
本指南不建議使用(yòng)任何特定的軟件生命周期模型或開發方法。申辦(bàn)方應建立适合其産(chǎn)品和組織的軟件生命周期模型,并滿足适用(yòng)的監管要求。所選的軟件生命周期模型應涵蓋軟件的整個産(chǎn)品生命周期。
01/文(wén)檔級别
Documentation Level
FDA打算采取基于風險的方法來幫助确定器械的文(wén)件級别,即Basic or Enhanced。文(wén)檔級别的目的是幫助确定軟件功能(néng)器械的上市前提交的最小(xiǎo)信息量。
器械的文(wén)件級别是基于其器械軟件功能(néng)在設備的預期用(yòng)途中(zhōng)的風險,因此文(wén)件級别反映了整個設備。
為(wèi)達到指南目的:
1.如果任何器械軟件功能(néng)的故障或缺陷可(kě)能(néng)會對患者、設備用(yòng)戶或使用(yòng)環境中(zhōng)的其他(tā)人造成可(kě)能(néng)導緻死亡或嚴重傷害的危險情況,則應對于器械軟件功能(néng)在内的任何上市前提交,提供Enhanced Documentation(增強文(wén)檔)。
在實施風險管路之前,應對這些風險進行評估。申辦(bàn)方應考慮設備預期用(yòng)途方面的風險(例如,對安(ān)全、治療和/或診斷的影響),以及其他(tā)相關考慮因素。
2.對于不适用(yòng)Enhanced Documentation(增強文(wén)檔)的器械軟件功能(néng)的上市前提交,應提供Basic Documentation(基本文(wén)檔)。
在确定Documentation Level(文(wén)檔級别)時,申辦(bàn)方應在實施風險管理(lǐ)之前考慮所有(yǒu)已知或可(kě)預見的軟件危害和與設備相關的危險情況,包括那些由合理(lǐ)可(kě)預見的誤用(yòng)引起的,無論是有(yǒu)意的還是無意的。這也包括器械網絡安(ān)全不足而有(yǒu)意或無意損害器械功能(néng)的可(kě)能(néng)性。
簡而言之,就是要申辦(bàn)方風險評估中(zhōng)積極、全面地考慮風險。
雖然在本指南範圍内的設備應單獨評估,以确定适當的Documentation Level(文(wén)檔級别),但對于某些類别的設備,FDA建議在上市前提交中(zhōng)提供Enhanced Documentation(增強文(wén)檔)。
如以下器械:用(yòng)于檢測因輸血傳播感染而獻血的器械、用(yòng)于确定獻血者和受血者相容性的器械、用(yòng)于采集供輸血或進一步生産(chǎn)使用(yòng)的血液和血液成分(fēn)的自動血細胞分(fēn)離機器械,以及血液機構的計算機軟件(BECS)。
除以上産(chǎn)品外,FDA認為(wèi),考慮到産(chǎn)品的性質(zhì)及其預期用(yòng)途,以下産(chǎn)品可(kě)能(néng)有(yǒu)獨特的風險,需要進一步的文(wén)件,以确保FDA能(néng)夠評估該器械的安(ān)全性和有(yǒu)效性。
包括組合器械(drug/device, biologic/device, drug/device/biologic)和III類器械。如果申辦(bàn)方評估Enhanced Documentation(增強文(wén)檔)級别不适合自身産(chǎn)品,那申辦(bàn)方必須提供适當的理(lǐ)由,說明為(wèi)什麽Basic Documentation(基本文(wén)檔)适合于上市前提交。在提交審查過程中(zhōng),如果需要評估器械的安(ān)全性和有(yǒu)效性,FDA可(kě)能(néng)會要求提供額外的信息。當然申辦(bàn)方也可(kě)以通過預審核與FDA進行溝通。
02/推薦文(wén)檔
Recommended Documentation
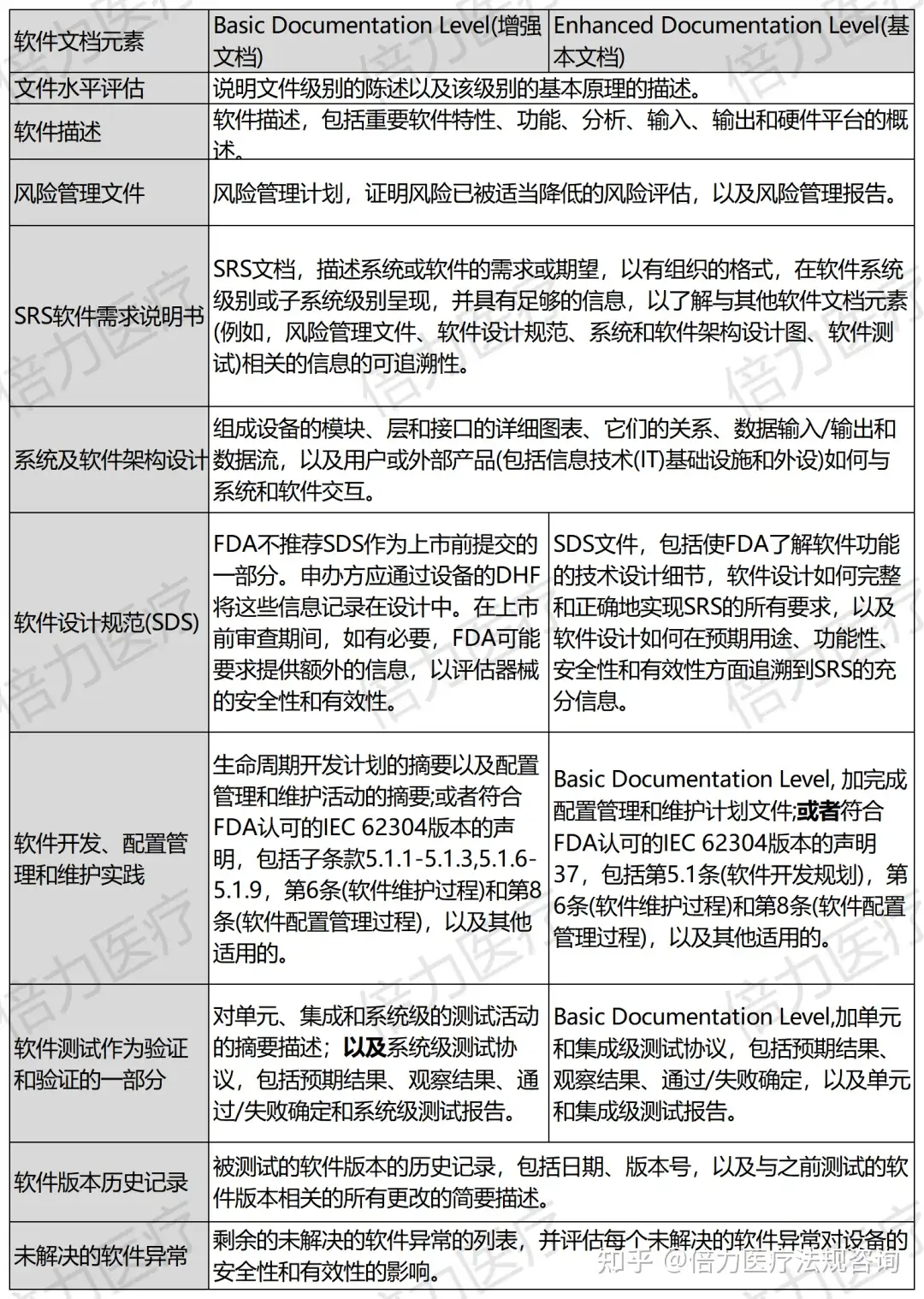
為(wèi)方便申辦(bàn)方更好的了解FDA對于Enhanced Documentation和Basic Documentation不同的要求,FDA也通過表格的形式進行對照,方便大家參考了解。
03/倍力總結
随着軟件發展叠代更新(xīn),FDA繼續更新(xīn)自己的監管措施,以順應時代的更叠。對于在2023年8月13日之前收到的提交文(wén)件的審查,通常CDRH工(gōng)作(zuò)人員不要求提供本指南中(zhōng)概述的新(xīn)的建議信息。然而,如提交了任何該最終指南中(zhōng)的此類信息,CDRH會進行審核。近日FDA也将舉辦(bàn)針對該最終指南的網絡研讨會。













